Empowered patients want greater sway over industry and the FDA
Print
02 March 2015
Marie Powers / BioWorld
Consumers are frustrated with the pace of drug and medical device development, and they want more engagement with industry and regulators at every step of the process, from trial design to FDA review. Savvy biopharmas and medical device manufacturers would be wise to heed that call by incorporating input from patients at every stage of development and sharing that feedback with the FDA as part of regulatory filings.
Those are just two conclusions in a thought-provoking report by Pwc's Health Research Institute that examined impressions from dozens of pharmaceutical and life sciences executives and hundreds of consumers about the pressure to deliver innovative and high-value products in what Pwc called the "new health economy."
In mid-2014, Pwc's Health Research Institute (HRI) surveyed 100 senior executives in the life sciences industry on issues such as regulation, the development of new therapies and patient engagement. Previous surveys were conducted in 1995, 1997, 1999, 2006 and 2010 as part of Pwc's Improving America's Health series.
Respondents to the 2014 survey represented a broad cross section of the pharmaceutical and life sciences industry in terms of head count, revenue and product lines, according to Pwc. In addition, 1,000 randomly selected adults in the U.S. were polled in an online survey about their views of the FDA, the pharmaceutical/life sciences industry and consumer access to therapies.
The resulting report, The FDA and industry: A recipe for collaborating in the New Health Economy, provides insights on the evolving relationship between industry and the FDA, the growing empowerment of consumers, the shift by payers from fee-for-service to value-based reimbursement models and the growing awareness by providers of the impact of cost in prescribing decisions. Each of these factors represents a key inflection point when evaluating the potential for a drug or device to be a legitimate game-changer.
For their part, industry executives said the FDA has become a more responsive partner and less of a "black box," according to Bobby Clark, senior manager at HRI. Seventy-eight percent of life sciences executives who responded to the poll indicated the agency improved the quality and frequency of its communications over the previous two years. Moreover, 76 percent said the FDA provides actionable feedback, and 70 percent said it offered more applicable guidance, rules and regulations than in years past.
But more can be done, survey participants added, with 71 percent indicating the FDA can accelerate approval programs by balancing swifter approval with increased postmarket surveillance.
"Executives really understand that balancing innovation and risk comes with tradeoffs," Clark said during a webinar sponsored by Pwc to discuss the report. "Executives are still eager to bring products to market as quickly as possible, but that's going to take some give and take in terms of how they work with regulators and consumers."
The regulatory framework for drugs and devices is "an evolving paradigm," added Michael Mentesana, pharmaceutical and life sciences R&D advisory services leader for Pwc. Since the FDA's establishment in 1906 following passage of the Pure Food and Drug Act, the agency has grappled with transforming from an organization primarily charged with protecting public health to one confronted with the need to advance innovation.
"That evolution is continual," Mentesana emphasized.
CONSUMERS 'SHOULDERING MORE AND MORE' COSTS
Industry attitudes also are shifting when it comes to defining value.
"We're seeing a little bit of warming in terms of recognition that products need to deliver clinical and economic value, particularly when it comes to dealing with purchasers and consumers, who are shouldering more and more of their treatment costs," Clark said. In fact, 43 percent of industry respondents endorsed the use of both economic and clinical value as a factor for drug approval, up from just 14 percent in 2010, representing a sea change in views.
But drugmakers also question whether they are getting the commensurate bang for their buck from the increased user fees flowing to the FDA. In 2013, those fees accounted for 57.9 percent of the budget for the FDA's Center for Drug Evaluation and Research (CDER), surpassing government funding to the agency for the first time, according to the Pwc report. In contrast, user fees represented only 25 percent of funding for the Center for Devices and Radiological Health.
"In the court of public opinion, that's an interesting statistic," Mentesana acknowledged, with fees rising from $23.1 million in 1993 to $718 million last year to CDER. "The big question out there is whether user fees and this FDA budget reliance have driven and fostered more innovation over time."
The fees have prompted a noticeable increase in staffing levels at the FDA, especially at CDER, as illustrated in Fig. 1 below. For the most part, review times also have improved, dropping from 13 months to 10 months for standard drug reviews and 13 months to seven months for premarket device approvals between 2008 and 2012. That said, standard drug review times were flat over the last three years of that time period. And while priority reviews for drugs dropped from 11 months to six months over the same period, they fluctuated wildly for medical devices, spiking from six months to 16 months between 2008 and 2009, then falling and spiking again before declining to eight months in 2012.
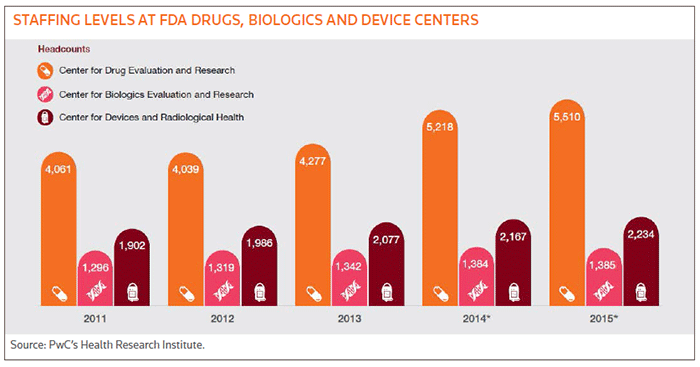
Just 32 percent of industry executives thought user fees resulted in shorter review times, and only 34 percent said the fees helped to accelerate innovation, according to Pwc. Part of the problem is that smaller biotechs, especially start-ups, have almost no visibility with the agency. More detailed and frequent communications with the FDA helps companies to understand and meet the agency's expectations during clinical development, so lack of access by smaller firms that represent the most innovative portion of the development cycle represents a huge disconnect for the industry, according to Pwc.
CONSUMERS 'WANT TO SEE THAT EFFICACY IS VERY HIGH'
Small firms also are the least likely to understand the nuances of the development pathways available for advancing promising therapies, including breakthrough therapy designation (BTD), accelerated approval, priority review and fast track designation, according to Chandresh Harjivan, a Pwc principal who leads management consulting within the firm's global public sector, which focuses on strategy, portfolio management and R&D in the public sector. Identifying and selecting the most appropriate pathway to approval is key from the get-go, since "companies that use these regulatory pathways can have many more meetings with the FDA," including informal gatherings that offer the opportunity for invaluable feedback.
Harjivan cited the example of Imbruvica (ibrutinib), which developer Pharmacyclics Inc. and partner Janssen Biotech Inc., a unit of Johnson & Johnson, moved to market in just four years using accelerated approval and BTD in a process he characterized as "lightning speed." Initial FDA approval in 2013 as a single agent to treat patients with mantle cell lymphoma who received at least one prior therapy has since been expanded to three additional indications. (See BioWorld Today, Nov. 14, 2013, and Jan. 30, 2015.)
As drug developers continue to look for the "secret sauce" in accelerating drug development, more skin in the game is prompting consumers to want a bigger say in the process. Three-fourths of consumer respondents to Pwc's survey said patients with life-threatening conditions should be able to access experimental therapies and indicated they would be willing to accept those risks personally, nearly matching the 77 percent of industry executives who agreed that such experimental therapies should be widely available.
Networking through advocacy groups and social media, "consumers are demanding the right to try unapproved treatments," Harjivan said. The "right to try can lead to expedited review," he added, posing a potential win-win for patients and biopharmas alike, with rigorous pharmacovigilance programs addressing regulator concerns.
"We've seen a lot of companies that get to market quickly have a much broader global view and can collect better data on those drugs with a larger group," Harjivan pointed out.
U.S. consumers also are looking more closely at the cost of new therapies. Although 49 percent of the Pwc survey respondents indicated they were willing to pay more for a "personalized medicine" product, 57 percent at some point declined to purchase an over-the-counter or prescription drug or device due to its price tag.
In part, heightened consumer awareness is a natural outcome of the fact that patients are paying more out of pocket. But today's consumers also are conducting their own research and making decisions on their health care in conjunction with their physicians rather than relying solely on the advice of medical authorities.
"Consumers are willing to pay to address a medical need, but they want to see that efficacy is very high, as well," Harjivan said.
Those providers also are evaluating cost as a component when prescribing drugs, with 54 percent responding in a 2014 HRI clinician survey that they consider cost "to a significant degree" when deciding whether or not to prescribe a drug.
How "the rise of the consumer" will affect the development and regulation of drugs in the second half of the decade remains to be seen, the Pwc officials conceded, but the FDA's move to create more patient-friendly labels is a likely first nod to that power, they said. (See BioWorld Today, Feb. 9, 2015.)
"Consumers are willing to provide information about their needs, their behaviors, their expectations and their treatments, but there's a next step that needs to be taken," Clark said, noting that patients want access to the right information and they want to know how to use the information. "They're moving from a lofty, abstract goal to more of a technical conversation about how to make that happen," he explained, so they can understand how to quantify and measure the risk associated with participating in the development of experimental therapies. "Companies and the FDA are beginning to think more about that," he added.
The 21st Century Cures initiative is a step in the right direction, according to Clark, but as consumers and patient advocacy groups become more engaged in the conversation, they want more than a seat on an FDA advisory committee.
"Those models are good, and they're important, but new models that add to those are needed," Clark said.
All Portfolio
MEDIA CENTER
-
The RMI group has completed sertain projects
The RMI Group has exited from the capital of portfolio companies:
Marinus Pharmaceuticals, Inc.,
Syndax Pharmaceuticals, Inc.,
Atea Pharmaceuticals, Inc.