Research primes pipeline with drug candidates aimed at CF
Print
05 April 2016
Peter Winter / BioWorld
In the U.S., about 1,000 new cases of cystic fibrosis (CF) – the rare, life-threatening genetic disease that causes persistent lung infections and progressively limits the ability to breathe – are diagnosed each year, according to the Cystic Fibrosis Foundation. While there are effective therapies that target the symptoms, there are no treatments that will fully restore lung function. However, genomic research is concentrating on a better understanding of the mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene in order to develop the next generation of therapies aimed at ameliorating the underlying genetic causes of CF.
Typifying that emerging trend is Boston-based Vertex Pharmaceuticals Inc., which strengthened its solid CF franchise by entering a major four-year strategic collaboration, option and license agreement with Basel, Switzerland-based company Crispr Therapeutics AG. (See BioWorld Today, Oct.27, 2015.)
GAME CHANGER
Vertex invested $105 million up front, including $75 million in cash and $30 million in equity, which gives it the option to an exclusive license for up to six CRISPR/Cas9-based treatments that emerge from the collaboration. CRISPR, or clustered regularly interspaced short palindromic repeats, and CRISPR-associated antigen Cas9 is known to act as the molecular scissors that cut and edit disease-associated DNA in a cell. That gene therapy/editing technology has been hailed as a potential "game changer" in a variety of diseases.
Vertex will fund all the discovery activities conducted under the agreement, which also encompasses other genetic diseases, including hemoglobinopathies such as sickle cell disease as well as CF. For each of the treatments in-licensed for development, Vertex will pay future development, regulatory and sales milestones of up to $420 million – potentially broadening the deal to $2.6 billion, plus royalties on product sales.
Initially, the companies will focus on the potential use of CRISPR/Cas9 to correct the mutations in the CFTR gene – known to result in the defective protein that causes CF.
With the deal, Vertex adds a potential gene therapy to its CF product pipeline, and David Altshuler, company chief scientific officer, noted that the collaboration could provide a "fundamental change in the future treatment of disease."
Certainly it is another string to the company's bow, which already includes two approved CF medicines: Orkambi (lumacaftor in combination with ivacaftor) – approved in the U.S. in July and in the EU in November, for the treatment of patients with CF, 12 years and older, who have two copies the F508del mutation in their CFTR gene; and Kalydeco (ivacaftor), which was approved in 2012 in the U.S. and the EU as a treatment for patients with CF, 6 years and older, who have the G551D mutation in their CFTR gene.
The company is also advancing two next-generation correctors – VX-152 and VX-440 – into clinical development. Those candidates are designed to improve processing and trafficking of the CFTR protein to the cell surface, beyond that observed with a single corrector combined with Kalydeco. (See BioWorld Today, Oct. 9, 2015.)
COMPETITION
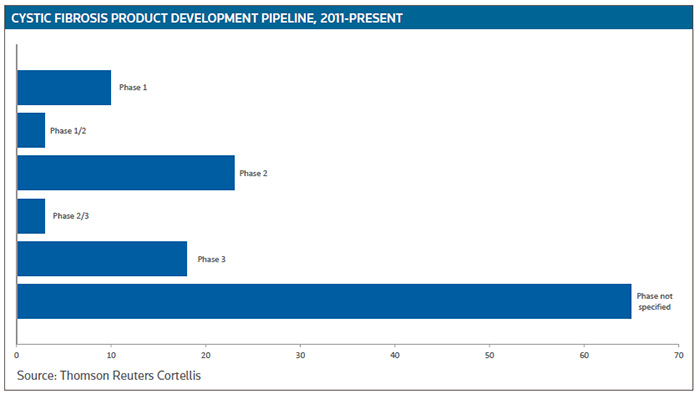
Vertex is doing everything it can to solidify its leading CF franchise because an increasing number of companies are involved in identifying and developing drug candidates for the treatment of CF. As a result, the product pipeline is expanding, with more than 40 ongoing mid- to late-stage clinical trials, according to Cortellis Clinical Trials Intelligence. (See Cystic Fibrosis Product Development Pipeline, below.)
This month, for example, Mechelen, Belgium-based Galapagos NV reported the first dosing of GLPG1837 in CF patients with the G551D mutation in the SAPHIRA 1 study. The drug candidate is a CFTR potentiator in development for the treatment of class III mutations CF. The full phase II program involves SAPHIRA 1, an open-label study of three doses of GLPG1837 in at least 12 patients with the G551D mutation. SAPHIRA 2 will be an open-label study of two doses of GLPG1837 in at least six CF patients with the S1251N mutation. Primary objectives are to evaluate the safety and tolerability, while secondary objectives are to assess changes in sweat chloride from baseline as the biomarker of CFTR ion channel function and to explore the changes in pulmonary function (forced expiratory volume in 1 second [FEV1]) from baseline. Both studies will include subjects treated with Kalydeco as well as those who are naïve to Kalydeco.
Nivalis Therapeutics Inc., of Boulder, Colo., meanwhile, has expanded its clinical development plan for lead investigational drug N91115, a first-in-class stabilizer of the CFTR protein, which is in an ongoing phase II study to evaluate its efficacy and safety in adult patients who have two copies of the F508del mutation and are being treated with Orkambi. Following positive review from an independent data monitoring committee (DMC) the study is now enrolling and randomizing patients in a higher (400 mg) dose cohort of N91115. The 12-week study is designed to compare two doses of N91115, 200 mg and 400 mg, administered twice daily when added to Orkambi.
Nivalis also said it will start a new trial in the second quarter to further evaluate the effect of N91115 in patients who have one copy of the F508del mutation and a second mutation that results in a gating defect in the CFTR protein. The phase II, proof-of-concept study will evaluate the efficacy and safety of N91115 in adult patients who have those mutations and who are being treated with Kalydeco.
N91115 works through a mechanism of action called S-nitrosoglutathione reductase (GSNOR) inhibition that is presumed to modulate the unstable and defective CFTR protein responsible for CF. GSNOR inhibition restores GSNO levels, thereby modifying the chaperones responsible for CFTR protein degradation. (See BioWorld Today, May 7, 2013.)
COMBATING INFLAMMATION
Laurent Pharmaceuticals Inc., of Montreal, is preparing for a phase II trial with LAU-7b, a once-a-day oral drug addressing the persistent and unresolved inflammation that represents a leading cause of lung tissue destruction in CF patients. The program has been enabled following the closing of a financing round led by Cystic Fibrosis Canada. LAU-7b is a solid dosage form of fenretinide that works, the company said, by correcting the defective metabolism of arachidonic acid (AA) and docosahexanoic acid (DHA).
"Although the impact of CFTR modulators on sweat chloride and lung function are exciting, they have not yet demonstrated an effect on inflammation," said Radu Pislariu, president and CEO of Laurent. "The AA/DHA imbalance in CF has long been recognized as a hallmark of the disease, but its potential in modulating the immune-inflammatory response in CF has been largely unexplored."
Corbus Pharmaceuticals Holdings Inc., of Norwood, Mass., is also targeting inflammation and has started patient enrollment in a phase II study of Resunab, an oral drug targeting the resolution of inflammation and fibrosis associated with disease progression in CF across all CFTR gene mutations. It will enroll about 70 adults with CF, irrespective of their CFTR mutation, to be treated daily for a period of 84 days, with a follow-up period of 28 days.
The company said it believes that because Resunab resolves inflammation independent of the underlying CF-related gene mutation or type of bacterial infection, it has the potential to improve the clinical outcome for a broad range of individuals with CF.
GENE THERAPY
Gene therapy solutions to CF have always been thought to be attainable, and results from a phase II trial last year may bear that out. The study, the results of which were published in Lancet Respiratory Medicine, was carried out by the UK Cystic Fibrosis Gene Therapy Consortium. It recruited 140 patients at 18 cystic fibrosis centers, and patients were randomized to receive either pGM169/GL67A gene liposome complex or saline, delivered once a month by a nebulizer.
Lung function was evaluated after one year using common clinical measure FEV1. In the 62 patients who received the gene therapy, FEV1 was 3.7 percent greater compared to placebo.
The trial is the first ever to show that repeated doses of a gene therapy can have a meaningful effect on the disease and change the lung function of patients. (See BioWorld Today, July 6, 2015.)
Proqr Therapeutics BV, of Leiden, the Netherlands, is taking an RNA-based approach, repairing the F508del mutation with its QR-010 so the messenger RNA can make CFTR protein with normal function.
The company has two global clinical studies of QR-010 ongoing. The first is a phase Ib study evaluating the safety, tolerability and pharmacokinetics of single and multiple ascending doses of inhaled QR-010 in 64 CF patients with the homozygous F508del mutation.
The second study is a proof-of-concept study evaluating the effect of QR-010 on an important measurement of CFTR function, the nasal potential difference (NPD). The company said it will enroll 16 CF patients, eight homozygous for the F508del mutation and eight compound heterozygous (one copy of the delta-F508 plus one other CF disease-causing mutation) with the option to enroll an additional 16. The NPD assay selectively measures the activity of the impaired CFTR protein in the nasal epithelium in CF patients. Restoration of NPD to normal levels will demonstrate an important proof of the effect of QR-010 on CFTR function, a benefit that has already been demonstrated in a mouse model with the F508del mutation. Top-line results for both studies are expected mid to late this year.
All Portfolio
MEDIA CENTER
-
The RMI group has completed sertain projects
The RMI Group has exited from the capital of portfolio companies:
Marinus Pharmaceuticals, Inc.,
Syndax Pharmaceuticals, Inc.,
Atea Pharmaceuticals, Inc.