Breakthrough designation: Cornucopia or gold star?
Print
02 September 2016
Brian Orelli / BioWorld
Since the FDA's breakthrough therapy designation came into existence as part of the FDA Safety and Innovation Act in July 2012, the agency has granted the designation to 145 drugs.
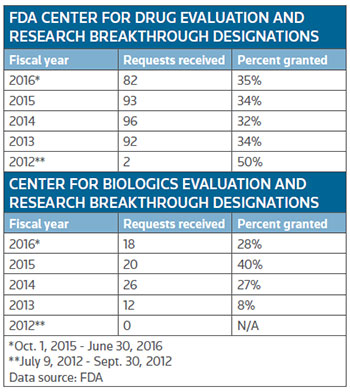 The Center for Drug Evaluation and Research (CEDR) has fairly consistently granted the designation to about a third of the applications, while companies applying to the Center for Biologics Evaluation and Research (CBER) haven't quite known what to expect with the designation granted to just 8 percent of the applications in the 2013 fiscal year, the first full year companies could apply, to a high of 40 percent granted in fiscal 2015. So far this fiscal year, through the month of June, CBER has granted the designation to 28 percent of applications. (See table, right.)
The Center for Drug Evaluation and Research (CEDR) has fairly consistently granted the designation to about a third of the applications, while companies applying to the Center for Biologics Evaluation and Research (CBER) haven't quite known what to expect with the designation granted to just 8 percent of the applications in the 2013 fiscal year, the first full year companies could apply, to a high of 40 percent granted in fiscal 2015. So far this fiscal year, through the month of June, CBER has granted the designation to 28 percent of applications. (See table, right.)
According to the FDA, the designation is for drugs that treat a serious or life-threatening disease or condition and where the "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints."
If the application is granted, which is supposed to be decided within 60 days of submitting the application, the FDA agrees to "expedite the development and review" of the drug, which includes all the features of the fast track program plus more intensive FDA guidance for the development of the most efficient clinical trials to speed development.
Not every company has decided that designation is helpful for its situation. Sarepta Therapeutics Inc., for example, has repeatedly said that it won't apply for the designation for its Duchenne muscular dystrophy drug eteplirsen. "We'd rather work through the process to determine the FDA's assessment of our clinical benefit, which is the criteria for breakthrough status," former CEO Chris Garabedian said on a conference call in October 2014.
Garabedian added that one of the main benefits of the designation – getting more interaction with the FDA – wasn't really an issue for Sarepta. "We believe we're getting the hands-on approach that [FDA Director of CEDR] Janet Woodcock has described with breakthrough designation. They have communicated as such publically of the level of attention they are giving to us."
EASIER DEVELOPMENT?
Stemline Therapeutics Inc., of New York, is the newest company to gain the FDA's breakthrough therapy designation for SL-401. The drug, which contains part of interleukin-3 fused to a toxic payload, was granted the designation for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN) last week.
Cells in BPDCN express high levels of the interleukin-3 receptor, making it a good initial indication to test SL-401. Stemline is also testing SL-401 in patients with acute myeloid leukemia, myeloproliferative neoplasms and multiple myeloma.
Results from an ongoing phase II clinical trial, most recently presented at the American Society of Clinical Oncology (ASCO) annual meeting and the 21st Congress of the European Hematology Association (EHA), both in June, showed that of the 19 BPDCN patients that could be evaluated, 17 had an overall response, with the two failures coming from patients that were already relapsed or refractory to an off-label drug. All 12 of the fist-line patients showed a response to the drug.
At the time, Stemline noted that it was still waiting for efficacy data from four additional patients who were enrolled in the clinical trial recently. Stemline is in ongoing discussions with the FDA about how much more data the company needs before it can file for regulatory approval.
"It's our position that we're there already," Ivan Bergstein, Stemline's president and CEO, told BioWorld Insight. "We think we've seen – continue to see – great data, and we'll see how those interactions go, so I'm sure by the end of the year we'll have a very clear sense of what they want to see."
After the designation was announced, analysts were quick to point out that it was a good sign that the FDA might be willing to allow Stemline to file a marketing application based on the company's current phase II data.
"With the breakthrough therapy designation, we consider it likely that the ongoing trial may serve as sufficiently registration-quality for SL-401 to receive accelerated approval in BPDCN without a need for any further efficacy trials," noted H.C.Wainwright and Co. analyst Raghuram Selvaraju.
Bergstein also thinks the designation is a good sign that the FDA may be willing to accept limited phase II data for a registration trial, "The breakthrough is a good first step in terms of getting them onboard."
A BioWorld Insight analysis of the 30 drugs with breakthrough therapy designation that were approved for their initial indication by CDER found that 13 of the drugs were given an accelerated approval by the FDA. Coincidently 13 drugs were approved based on phase II or earlier clinical trial data alone although some of those were actually full approvals, typically for orphan indications.
Looking specifically at the 15 drugs that were approved for cancer indications, 12, or 80 percent, were given accelerated approval and 10 of the 15 drugs were approved based on phase II or earlier clinical trial data.
Another potentially good sign for Stemline is that the designation includes all stages of BPDCN, while most oncology drugs are given breakthrough therapy designation for a subset of patients with the type of cancer as highlighted by Roth Capital Parners analyst Joseph Pantginis in a note to clients: "This is unique compared to other therapies that are granted the designation based on a particular subgroup or stage of disease."
For instance, last month, Darzalex (daratumumab, Genmab A/S/Janssen Biotech Inc.) was given breakthrough therapy designation for patients with multiple myeloma who have received at least one prior therapy. And Opdivo (nivolumab, Bristol Myers Squibb Co.) received the designation in June for urothelial carcinoma was specifically for unresectable locally advanced or metastatic urothelial carcinoma that has progressed on or after a platinum-containing regimen.
Getting the designation for all patients with BPDCN highlights the severe unmet medical need for BPDCN Bergstein pointed out, "It indicates that the FDA appreciates that the disease universally is bad whether you're untreated or treated – it goes from bad to worse – and I think that's a testament to why we got the broad BPDCN indication."
All Portfolio
MEDIA CENTER
-
The RMI group has completed sertain projects
The RMI Group has exited from the capital of portfolio companies:
Marinus Pharmaceuticals, Inc.,
Syndax Pharmaceuticals, Inc.,
Atea Pharmaceuticals, Inc.